




La surveillance des encéphalopathies subaiguës spongiformes transmissibles (ESST) en France, données 2009-2010
// Surveillance of human transmissible spongiform encephalopathies (TSEs) in France in 2009 and 2010
Résumé
Introduction –
Le Réseau national de surveillance des maladies de Creutzfeldt-Jakob (RNS-MCJ) repose sur une organisation nationale et pluridisciplinaire. Son objectif est de détecter tous les cas d’encéphalopathies subaiguës spongiformes transmissibles (ESST) humaines afin de les classer par étiologie, d’en estimer l’incidence, de décrire leur répartition temporo-spatiale et leurs tendances évolutives, et de détecter les cas groupés.
Matériels et méthodes –
L’analyse descriptive a porté sur les données recueillies au RNS-MCJ en 2009 et 2010 en France métropolitaine et dans les départements et territoires d’outre-mer.
Résultats –
Au total, 1 485 et 1 614 suspicions d’ESST ont été signalées au RNS-MCJ en 2009 et 2010. Un diagnostic final d’ESST a été retenu au décès pour 312 cas. Ces cas étaient répartis sur tout le territoire sans regroupement. Deux cas de la variante de la MCJ (vMCJ) sont décédés en 2009. La distribution des cas dans les différentes classes d’ESST est similaire à celle observée les années précédentes et les cas présentaient des caractéristiques identiques en termes d’âge, de tableau clinique et de tableau lésionnel.
Conclusion –
Le suivi de l’évolution de l’épidémie de vMCJ et la possible émergence de nouvelles formes zoonotiques justifient le maintien de la surveillance des ESST.
Abstract
Introduction –
The French national network of Creutzfeldt-Jakob diseases surveillance (RNS-MCJ) relies on a national and multidisciplinary organization. It aims to detect all forms of human transmissible spongiform encephalopathies (TSEs) in order to classify them, to estimate their incidence, to describe their spatiotemporal distribution and to detect the case clusters.
Material and methods –
The descriptive analysis used the data collected by the RNS-MCJ in 2009 and 2010 in metropolitan France and in the overseas departments and territories.
Results –
In 2009 and 2010, 1,485 and 1,614 suspicions of TSEs were notified to the RNS-MCJ. A final diagnosis of TSE was retained at death for 312 cases. No cluster was detected. Two cases of variant CJD died in 2009. The distribution of age, clinical and neuropathological characteristics and forms of TSE were similar to those observed previous years.
Conclusion –
Monitoring of vCJD epidemiology and the possible emergence of new zoonotic forms support the need for sustained active surveillance.
Introduction
Les encéphalopathies subaiguës spongiformes transmissibles (ESST), ou maladies à prions, sont des affections neurodégénératives se développant aussi bien chez l’homme que l’animal et ayant comme caractéristique d’être transmissibles et dues à un agent transmissible non conventionnel (ATNC), le prion (proteinaceous infectious particle). Selon l’hypothèse du prion, l’agent transmissible serait constitué d’agrégats d’une forme anormale d’une protéine de l’hôte, la PrP, codée par le gène PRNP chez l’homme. La PrP existe sous deux formes : la PrPc, forme cellulaire, normale et sensible aux protéases, ancrée à la membrane des cellules de nombreux tissus, en particulier les neurones, et la PrPsc, forme pathologique résistante aux protéases. La PrPsc se différencie de la PrPc par sa conformation acquise selon un mécanisme post-traductionnel lui conférant ses propriétés de résistance aux protéases et d’agrégation. Dans toutes les formes d’ESST, l’accumulation de PrPsc dans le système nerveux central (SNC) est retrouvée.
La transmission des ESST est possible au sein d’une même espèce animale mais plus difficile d’une espèce à l’autre, en fonction notamment du degré d’homologie de séquence des gènes codant la PrP du donneur et du receveur.
Les ESST se caractérisent par leur rareté, leur longue durée d’incubation, leur évolution fatale sans rémission et sans aucune réaction inflammatoire ou immunitaire détectable, et par un tableau neuropathologique particulier associant des lésions de spongiose, de gliose, de raréfaction neuronale et des dépôts extracellulaires amyloïdes (ou non) de protéine prion pathologique 1.
Chez l’homme, on distingue la maladie de Creutzfeldt-Jakob (MCJ) sporadique, les formes génétiques et les formes acquises.
La MCJ sporadique touche des patients âgés en moyenne de 65 ans. Après une phase de prodromes aspécifiques, une démence rapidement évolutive s’installe, diversement associée à des signes neurologiques (myoclonies, syndrome cérébelleux, troubles visuels, syndrome pyramidal et extrapyramidal). L’origine de la forme sporadique de la MCJ n’est pas connue. Plusieurs études épidémiologiques de type cas-témoins ont été réalisées mais n’ont pas permis d’identifier de facteur pouvant expliquer l’ensemble des cas de MCJ sporadiques 1. Néanmoins, il existe chez l’homme un polymorphisme sur le codon 129 du gène PRNP, définissant trois génotypes possibles : méthionine/méthionine (MM), valine/valine (VV) ou méthionine/valine (MV). Dans la population générale, on observe 50% d’homozygotes (MM ou VV) et 50% d’hétérozygotes (MV). Dans la MCJ sporadique, 80% des patients sont homozygotes avec une nette prédominance du génotype MM 2. L’homozygotie MM constitue donc un facteur de risque pour la MCJ sporadique mais non un facteur causal 1.
Les formes génétiques des ESST sont liées à l’existence de mutations ou d’insertions sur le gène PRNP. Elles se transmettent sur le mode autosomique dominant avec une pénétrance variable. En fonction du type de mutation, du tableau clinique et des données neuropathologiques, on différencie les MCJ génétiques, le syndrome de Gerstmann-Straüssler-Scheinker (SGSS) et l’insomnie fatale familiale (IFF) 3.
Les formes acquises regroupent le Kuru, décrit uniquement en Papouasie-Nouvelle-Guinée, les MCJ iatrogènes et la variante de la MCJ (vMCJ). Les MCJ iatrogènes résultent d’une contamination survenant lors de l’injection sous-cutanée ou intramusculaire d’hormone de croissance d’origine humaine, au cours de greffes de tissus (cornée, dure-mère) ou à la suite de l’utilisation d’instruments neurochirurgicaux insuffisamment décontaminés.
La vMCJ, seule forme liée à l’encéphalopathie spongiforme bovine (ESB), touche principalement l’adulte jeune (autour de 35 ans en France). Les signes inauguraux sont souvent d’ordre psychiatrique (dépression, état délirant, illusions, hallucinations), associés parfois à des douleurs. Après quelques mois, les signes neurologiques apparaissent (principalement ataxie et myoclonies). Enfin, une démence s’installe progressivement avec une évolution finale analogue à la MCJ sporadique. Tous les cas observés à ce jour sont MM au codon 129 3.
En 1992, un réseau d’épidémiosurveillance de la MCJ a été mis en place par l’Institut national de la santé et de la recherche médicale (Inserm) en raison du risque de passage de l’agent de l’ESB à l’homme et de l’apparition des cas de MCJ liés à l’hormone de croissance.
Avec l’émergence de la vMCJ en 1996, puis la confirmation que cette nouvelle forme de MCJ résultait du passage de l’agent de l’ESB à l’homme, la MCJ et les autres ESST humaines ont été inscrites sur la liste des maladies à déclaration obligatoire (DO).
À la fin de l’année 2000, après la deuxième « crise de la vache folle », le Réseau national de surveillance des maladies de Creutzfeldt-Jakob et des maladies apparentées (RNS-MCJ), structure pluridisciplinaire, a été créé. Il a pour objectif principal de détecter tous les cas d’ESST humaines, et plus particulièrement les cas de vMCJ et de MCJ iatrogènes, afin de les classer par étiologie, d’en estimer l'incidence, de décrire leur répartition temporo-spatiale et leurs tendances évolutives, et de détecter les cas groupés.
Les objectifs de cet article sont de décrire les signalements d’ESST déclarés au RNS-MCJ en 2009 et 2010, d’évaluer la performance du RNS-MCJ et les validités intrinsèques du test de recherche de la protéine 14-3-3, de l’EEG et de l’IRM.
Matériels et méthodes
Le système de surveillance
Le RNS-MCJ est coordonné par l’équipe maladie d'Alzheimer-maladies à prions de l’Inserm UMR-S 975. Il comprend les équipes suivantes :
- les services de biologie de l’Hôpital Lariboisière à Paris (AP-HP), de l’Hôpital neurologique de Lyon (Hospices civils) et des CHU de Bordeaux, Montpellier et Reims ;
- le Centre national de référence des agents transmissibles non conventionnels ;
- la Cellule nationale de référence des MCJ ;
- le Réseau de neuropathologie.
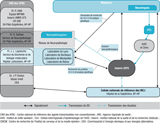
Les données épidémiologiques, cliniques, génétiques et neuropathologiques sont recueillies selon des protocoles standardisés. Il existe quatre possibilités de notification au RNS-MCJ (figure 1) :
- le signalement de tests de détection de la protéine 14-3-3 par les laboratoires de biologie du RNS-MCJ (majorité des cas). La protéine 14-3-3 est une protéine ubiquitaire particulièrement abondante dans les neurones et régulatrice de nombreuses fonctions cellulaires. Sa présence dans le liquide céphalo-rachidien est un indicateur de souffrance neuronale. Cette protéine a une valeur diagnostique importante pour les ESST et notamment pour la MCJ sporadique ;
- la DO de cas suspects par les praticiens à leur Agence régionale de santé (ARS) ;
- les praticiens qui contactent directement le RNS-MCJ ou la cellule nationale de référence des MCJ (qui transmet la notification au RNS-MCJ) ;
- le Réseau de neuropathologie, qui signale directement des cas d’ESST.
 Agrandir l'image
Agrandir l'image
Les signalements reçus sont centralisés à l’UMR S-975 et revus individuellement pour vérifier s’ils répondent aux critères diagnostiques des différentes formes d’ESST établis par l’European Creutzfeldt-Jakob Diseases Surveillance Network (EuroCJD) (tableau 1). Seuls les cas probables et certains sont publiés mensuellement sur le site de l’Institut de veille sanitaire (InVS). Les cas possibles ne sont pas comptabilisés en raison d’une trop grande incertitude diagnostique. Un cas défini ou certain est un patient pour lequel le diagnostic d’ESST a été confirmé après le décès du patient par un examen neuropathologique et immunocytochimique du cerveau. Les cas probables et possibles sont définis selon un faisceau de critères cliniques (signes à l’examen neurologique) et paracliniques (exploration IRM, EEG et recherche de la protéine 14-3-3). Chaque cas est suivi jusqu’à l’obtention d’un diagnostic final (ESST ou autre diagnostic) et d’un classement étiologique (MCJ sporadique, ESST génétique, MCJ iatrogène ou vMCJ) (figure 1, tableau 1)
 Agrandir l'image
Agrandir l'image
Analyse des données
L’analyse des données a été réalisée à l’aide du logiciel Stata® 12.1 (StataCorp. 2011. Stata Statistical Software: Release 12. College Station, TX: StataCorp LP).
La répartition spatiale et temporelle ainsi que les caractéristiques démographiques et cliniques des cas certains, probables et possibles d’ESST toutes formes confondues, décédés en 2009 et en 2010, ont été décrites.
La performance du réseau de surveillance a été évaluée par l’analyse du délai de signalement (délai entre la date de début des signes et le signalement au RNS-MCJ) et par la proportion des cas ayant fait l’objet d’un examen neuropathologique. La performance de la DO a été évaluée par son exhaustivité et par le pourcentage de cas probables et certains d’ESST.
Nous avons évalué la validité intrinsèque (sensibilité et spécificité) du test de recherche de la protéine 14-3-3, de l’EEG et de l’IRM à partir des cas certains de MCJ sporadiques et des suspicions pour lesquelles un diagnostic différentiel a été posé en 2009 et 2010. Pour l’IRM, le calcul de sensibilité et de spécificité a porté sur les cas certains de MCJ sporadiques et les diagnostics différentiels rapportés au RNS-MCJ en 2010 uniquement, cet examen n’ayant été inclus dans les critères de diagnostic positif de MCJ qu’à partir de 2010.
La présence de la protéine 14-3-3 peut être contrôlée jusqu’à quatre fois pour un même patient. Elle est considérée comme positive lorsqu’au moins une recherche de la protéine est positive. La valeur manquante est attribuée lorsqu’aucun résultat de recherche n’a été enregistré. La recherche de la protéine 14-3-3 est négative dans tous les autres cas.
L’EEG est positif lorsqu’il est périodique ou pseudopériodique. La valeur manquante a été attribuée aux cas dont l’EEG n’avait pas été réalisé ou était ininterprétable. L’EEG est négatif pour les modalités : normal, ralenti et non caractéristique.
L’IRM est positive lorsque des hypersignaux dans le noyau caudé et/ou le putamen ont été identifiés. La valeur manquante a été attribuée quand l’IRM n’a pas été réalisée ou a été effectuée avant 2010. Autrement, l’IRM est considérée comme négative.
Résultats
Nombre de suspicions d’ESST signalées au RNS-MCJ en 2009 et 2010
En 2009 et 2010, 1 485 et 1 614 suspicions d’ESST ont été signalées au RNS-MCJ. Ce nombre de suspicions a augmenté de 9% entre 2008 et 2010 (figure 2). En 2009 et 2010, respectivement 146 et 166 cas d’ESST ont été retenus sur la base de l’évaluation finale après leur décès et de leur classement comme forme possible, probable ou certaine. La suite de l’analyse porte sur ces 312 cas.
 Agrandir l'image
Agrandir l'image
Descriptions des caractéristiques des cas d’ESST décédés en 2009 et 2010
Parmi les 312 cas d’ESST décédés en 2009 et 2010, ont été diagnostiqués : 282 cas de MCJ sporadiques certains, probables et possibles, 2 cas de vMCJ (1 probable et 1 certain), 4 cas de MCJ iatrogènes et 24 cas d’ESST de formes génétiques (tableau 2).
 Agrandir l'image
Agrandir l'image
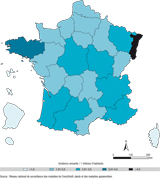
Les cas observés de MCJ sporadiques certains et probables décédés en 2009-2010 résidaient dans toutes les régions françaises (figure 3). L’incidence de la MCJ sporadique en France calculée sur les deux années (2009 et 2010) était de 2,54 cas par million d’habitants, avec des incidences extrêmes de 4,05 et 3,13 cas par million d’habitant respectivement pour l’Alsace et la Bretagne.
 Agrandir l'image
Agrandir l'image
La proportion de femmes atteintes par une ESST était plus importante que celle des hommes (sex-ratio H/F=0,77).
L’âge moyen au décès de l’ensemble des cas était de 69 ans ; il était significativement différent selon les différentes formes d’ESST. Il était de 70 ans (extrêmes : 48-89 ans) pour les MCJ sporadiques certaines et probables ; de 58 ans pour les MCJ génétiques et de 35 ans pour les MCJ iatrogènes liées à l’hormone de croissance. Le cas certain de vMCJ avait 53 ans au moment du décès, le cas probable 45 ans.
En termes de caractéristiques cliniques à la fin de la maladie, 97% des patients, toutes formes d’ESST confondues, présentaient des troubles intellectuels, 75% des myoclonies et 63% un syndrome cérébelleux. Parmi les cas de MCJ sporadiques, ces signes cliniques étaient respectivement retrouvés chez 97%, 77% et 65% des patients.
L’analyse du gène PRNP a été effectuée pour 208 cas parmi les 312 cas d’ESST décédés en 2009 et 2010 (67%). Le génotype MM au codon 129 était le plus fréquent (54%) (tableau 3).
 Agrandir l'image
Agrandir l'image
Les MCJ génétiques étaient liées à des mutations E200K (13 cas), V210I (3 cas), E211Q (1 cas), et D178N (1 cas). Un cas de MCJ génétique était lié à une insertion de 168 paires de bases. Sur les deux cas de SGSS, l’un était lié à une mutation du codon 102 et l’autre à une insertion de 192 paires de bases. Les deux cas d’IFF portaient une mutation du codon 178, associée à la présence d’une méthionine sur l’allèle muté. La notion d’antécédents familiaux était connue pour les 3 cas d’IFF. Cette notion était retrouvée chez 9 cas de MCJ génétiques (47%).
Performance du système de surveillance
En 2009 et 2010, 150 patients ont eu un examen neuropathologique, qui a permis d’identifier 113 cas d’ESST et 37 diagnostics différentiels. Parmi ces 37 derniers patients, 14 répondaient aux critères de MCJ sporadique avant l’autopsie, 7 étaient classés MCJ probables et 7 MCJ possibles.
Le nombre de cas classés sporadiques probables, c’est-à-dire non autopsiés, reste nettement supérieur au nombre de cas confirmés par l’examen neuropathologique (163 cas contre 102 parmi les 265 cas de MCJ sporadiques observés en 2009 et 2010).
Sur les 312 cas d’ESST, 169 des (54%) ont fait l’objet d’une DO à l’ARS, dont 62 cas certains, 100 cas probables et 7 cas possibles. En revanche, 16 patients ayant fait l’objet d’une DO de suspicion d’ESST n’étaient pas atteints d’ESST.
Le délai de signalement (extrêmes : 0-83 mois) était inférieur ou égal à 6 mois dans 86% des cas. Le délai maximal enregistré concernait un cas de MCJ sporadique certain signalé après le décès du patient par le Réseau de neuropathologie.
La sensibilité du test de détection de la protéine 14-3-3 a été estimée à 86% et sa spécificité à 92%. La sensibilité et la spécificité étaient respectivement de 49% et 78% pour l’EEG et de 68% et 91% pour l’IRM.
En 2010, parmi les cas certains, probables ou possibles d’ESST, 81% (134 sur 166) patients ont bénéficié à la fois de la recherche de la protéine 14-3-3 et de l’IRM, et 31% avaient une IRM positive associée à une recherche négative de la protéine 14-3-3 ou une IRM négative associée à une recherche positive de la protéine 14-3-3.
Discussion
Le nombre de suspicions d’ESST signalées a continué à augmenter pour les années 2009 et 2010, pour un nombre de cas restant globalement stable. L’incidence de la MCJ sporadique observée en France de 1993 à 2010 est de 1,44 cas par million d’habitants (extrêmes : 0,59-2,26), et de 1,48 et 1,50 respectivement pour l’Alsace et Bretagne. Il est donc probable que les incidences extrêmes observées en 2009 et 2010 dans ces deux régions ne sont que le reflet de fluctuations liées au hasard. Bien entendu, l’incidence de ces deux régions sera particulièrement surveillée dans les prochaines années. La répartition des cas dans les différentes classes d’ESST est similaire à celle observée les années précédentes. L’analyse du gène PRNP a permis d’identifier 23 cas d’ESST génétiques, tous porteurs de mutations déjà décrites. Avec 2 cas décédés en 2009, le nombre cumulé de cas vMCJ probables et certains, en France, s’élevait alors à 25.
La moitié des cas d’ESST probables et certains ont fait l’objet d’une DO à l’ARS. Dans l’intérêt du patient et de sa famille, il est important de rappeler aux médecins la nécessité de cette déclaration, puisque celle-ci est l’unique déclencheur de la mise en place d’une aide financière spécifique aux MCJ (aide d’urgence prévue dans la circulaire DGH /DHOS/DGAS/DSS n°2001-139 du 14 mars 2001).
La proportion d’autopsies réalisées entre 2009 et 2010 (37%) est plus faible que celle observée durant la première décennie de la mise en place du RNS-MCJ, 58% en moyenne, avec un pic à 64% en 2000. Il reste donc important d’encourager la réalisation de l’examen neuropathologique, indispensable au diagnostic de certitude des ESST.
L’étude de la sensibilité et de la spécificité de la recherche de la protéine 14-3-3 et de l’IRM retrouve des performances comparables à celles observées dans la littérature 4,5.
Le suivi de l’évolution de l’épidémie de vMCJ et la possible émergence de nouvelles formes zoonotiques justifient le maintien de la surveillance des ESST 6.
Remerciements
Nous remercions l’ensemble des acteurs du RNS-MCJ.